6. Wichtige Erkrankungen mit assoziierten Nahrungsmittelunverträglichkeiten
Autoren: M. Raithel, A. Hagel, W. Taumann, P.Konturek, G.J. Molderings, U. Hetterich
6.1 Reizdarm ( auch Reizdarmsyndrom / Irritable Bowel Syndrome / IBS / RDS)
Beim Reizmagen oder Reizdarm werden häufig Nahrungsprobleme, postprandiale (nach der Nahrungsaufnahme) Symptome oder Körperbeobachtungen nach dem Essen berichtet. Daher wird in dieser Patientengruppe oft das Vorliegen einer Nahrungsmittelunverträglichkeit bzw. Nahrungsmittelallergie vermutet (1–3). Es ist bekannt, dass unter der Diagnose Reizdarmsyndrom gehäuft Patienten zu finden sind, bei denen eine der oben aufgeführten Erkrankungen vorliegt, aber noch nicht erkannt ist, weil vielleicht ein klinisch inapparenter (nicht in Erscheinung tretender), asymptomatischer oder atypischer Verlauf vorliegt. Dies gilt insbesondere für die relativ hohe Koinzidenz des Reizdarmsyndroms mit zum Beispiel der Kohlenhydratmalassimilation, NMA, NMU und der Zöliakie (1–3, 11, 20–25).
Andererseits finden sich aber auch Reizdarmpatienten, bei denen die Genese der Beschwerden trotz umfangreicher Differenzialdiagnostik noch nicht geklärt werden kann und bislang als idiopathisch (ohne erkennbare Ursache), neurovegetativ oder psychosomatisch einzustufen sind. Daher ist auch eine entsprechende psychosomatische Konsiliaruntersuchung mindestens einmal in einer Patientenkarriere durchzuführen.
Mit den in der Abbildung 3 dargestellten Diagnostikmodalitäten sollte daher stets versucht werden, die Differenzialdiagnose des Reizdarms soweit wie möglich zu klären bzw. den Nachweis einer anderen Erkrankung zu führen, wenn rezidivierend (wiederkehrende) Nahrungsmittelunverträglichkeiten geklagt werden.
Interessanterweise konnte bei einer Studie an Reizdarmpatienten festgestellt werden, dass, bei der überwiegende Anzahl der Patienten, deutlich überaktivierte Mastzellen zu finden sind, welche durch sekundäre Unverträglichkeitsreaktionen oder primäre genetische Mutationen verursacht sein können (48).
6.2 Mastzellerkrankungen
„Systemische Mastzellaktivierungserkrankung“ (MCAD für mast cell activation disease) ist der Überbegriff für eine Gruppe von Erkrankungen, nämlich der systemischen Mastozytose (SM) mit ihren Subtypen, dem Mastzellaktivierungssyndrom (MCAS, mast cell activation syndrome) und der Mastzellleukämie (MCL) (siehe Abbildung 5). Molekulare Ursache der Erkrankung sind multiple genetische Veränderungen in Kinasen, Rezeptoren und anderen Proteinen der intrazellulären Signalverarbeitung (sog. Signaltransduktionskette) in einem Teil der Mastzellen, die zu einer konstitutiven Aktivitätssteigerung von betroffenen Mastzellen führen (49). Der unregulierte, erhöhte Aktivitätszustand geht mit einem gestörten Apoptose- und – bei bestimmten Mutationen – auch erhöhtem Proliferationsverhalten einher mit einer langsamen Anreicherung dieser krankhaft veränderten überaktiven Mastzellen in Organen und Geweben. Die unkontrolliert freigesetzten Mediatoren aus den in den Organen und Geweben vorhandenen pathologischen Mastzellen und vor allem aus durch diese kaskadenhaft sekundär aktivierten gesunden Mastzellen gelangen in das Gewebe und verursachen dort gewebetypische Veränderungen und lokale Symptome, die oft anfangs nicht erkannt werden, da derartige Erhöhungen der Gewebemediatoren in der klinischen Routine in der Regel nicht erfasst werden. Bei Übertritt von Mediatoren in die Blutbahn oder bei deren Transport über die Lymphe können systemische Beschwerden direkt über Stimulation von Erfolgszellen und wahrscheinlich indirekt durch Stimulation von residenten Mastzellen in anderen Geweben und Organen induziert werden (49). Abhängig von den betroffenen Geweben und Organen und den daraus resultierenden für den Patienten dominierenden Beschwerden kann sich die Erkrankung in verschiedenen klinischen Varianten präsentieren, z.B. Reizdarmsymptomatik (Durchfall/Verstopfung, Bauchkrämpfe, Gastritis, Enteritis, Colitis, eher untergewichtig, episodisch oder dauerhaft hypoton), Fibromyalgiesymptomatik (rheumaartige Gelenk- und Muskelschmerzen, brennende Schmerzen in wechselnden Hautarealen, Parästhesien), Kardialer Phänotyp (Tachyarrhythmien mit/ohne Palpitationen, nicht-kardiale thorakale Schmerzen, Dyspnoegefühl), ZNS-Phänotyp (Schwindel, episodisch oder dauerhaft hyperton, eher übergewichtig, Tinnitus, Wortfindungsstörungen, Konzentrationsstörungen, Schlaflosigkeit, Panikattacken, depressive Zustände, chronische Müdigkeitssymptomatik, Hyperventilationstetanie-ähnliche Zustände), „Idiopathische“ Anaphylaxie (dermale, respiratorische, kardiovaskuläre, intestinale Symptome) (49).
Bis vor kurzem war es weitverbreitete Ansicht, dass die MCAD zu den seltenen Erkrankungen gehört. Dies trifft allerdings nur für zwei Unterklassen zu: für die systemische Mastozytose und die Mastzellleukämie, deren Prävalenzen mit 1:364 000 bzw. noch mindestens 2 Größenordnungen geringer berechnet wurden (49). Dagegen betrug die Prävalenz der MCAD in der deutschen Bevölkerung in einer aktuellen ersten Studie bis zu 17 % (49). Diese hohe Prävalenz muss in weiteren großen epidemiologischen Untersuchungen noch bestätigt werden, dennoch wird eine mittlere Prävalenz von 5 – 10% geschätzt. Überraschend ist diese hohe Prävalenz jedoch nicht, denn zur Aktivierung der Mastzellen können zudem primäre und sekundäre Prozesse (andere Krankheitsmechanismen) beitragen, genetisch prädisponierende Situationen werden zunehmend häufiger erkannt und nicht zuletzt wird das Auftreten des Mastzellaktivierungssyndroms auch bei verschiedenen Erkrankungsbildern wie z.B. bei bestimmten Formen der Fibromyalgie, Untergruppen des Reizdarmsyndroms, rheumatischen Erkrankungen und Autismus (49, 50) beobachtet.

Anders als bisher in den WHO-Kriterien für Mastozytose angenommen, für welche das Vorliegen der C-KIT Punktmutation KIT-Punktmutation in Codon 816 in Mastzellen aus dem Knochenmark oder aus einem anderen, extrakutanen Organ ein Hauptkriterium ist, sind bereits eine Vielzahl von weiteren Mutationen im C-KIT Gen sowie in einer Vielzahl von weiteren Genen, z.B. PDGFR, CBL, TET2, ASXL1, JAK2, U2AF1, SETBP1, RUNX1, DNMT3, als ursächlich mitbeteiligt für eine Mastzellerkrankung identifiziert worden (50, 52). Durch die starke familiäre Anhäufung von Mastzellerkrankungen wird weiterhin eine epi-genetische Komponente vermutet (51). Zusammenfassend ist die systemische Mastzellerkrankung eine polygene und epi-genetische Erkrankung, deren Grundursache vermutlich in individuell unterschiedlichen Kombinationen von insbesondere somatischen und germline Mutationen in den protein-kodierenden Genen des RNA-Splicing-Mechanismus, den protein-kodierenden Genen der Transkript-Steuerung und in den Epigenetik-regulierenden Genen liegt (50, 52).
Für die komplexe Diagnose, z.B. Anamnese hinsichtlich eines Mastzellmediatorsyndroms mit Hilfe eines standardisierten validierten Fragebogens, Mediatorbestimmung (Tryptase, Heparin im Blut, Methylhistamin im Urin) oder Gastro-/Koloskopie mit Biopsien und mastzellspezifische immunhistochemische Untersuchung der Gewebeprobe, als auch die Therapie, z.B. beginnend mit einer Basistherapie (H1-Antihistaminikum, Cromoglicinsäure, Ranitidin, Vitamin C 500 mg retard Kapseln), stehen neu überarbeitete CME-zertifizierte Leitlinien zur Verfügung (49).
Mastzellerkrankungen führen zu zahlreichen Nahrungsmittelunverträglichkeiten und Allergien, die, je Patient, individuell stark variieren können. Daher ist eine generelle Ernährungsempfehlung schwierig und für mastzellerkrankte Patienten empfiehlt sich eine Stufen- bzw. Ausschlussdiät, um den optimalen Ernährungsplan zu erarbeiten. Unterschätzt werden oft die Auswirkungen der durch aktivierte Mastzellen ausgeschütteten Mediatoren und Zytokine auf das zentrale Nervensystem (ZNS) mit häufiger Konsequenz von somatoformen Störungen, die in der Regel als rein exogene psychisch-bedingte Störungen fehlinterpretiert werden (siehe Abbildung 9).
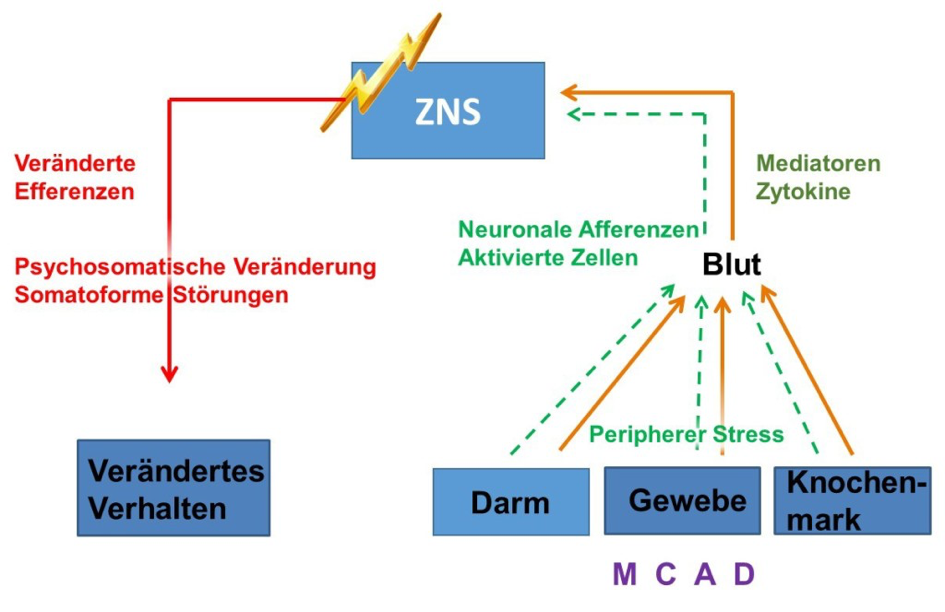
Durch die zentrale Rolle der Mastzellen bei Nahrungsmittelallergien und Nahrungsmittelunverträglichkeiten kann erwartet werden, dass die zunehmend intensive Erforschung der Funktionsweise der Mastzelle auf verbesserten Diagnose- und Therapiemöglichkeiten bei Nahrungsmittelallergien und Nahrungsmittelunverträglichkeiten führen wird.
6.3 Chronisch-entzündliche Darmerkrankungen und mikroskopische Enterocolitiden
Weitere organische Erkrankungen des Magen- Darmtraktes, die mit einer Störung der Nahrungsresorption oder Nahrungstoleranz einhergehen und in der Diagnostik der Nahrungsmittelunverträglichkeit und Nahrungsmittelallergie eine Rolle spielen, sind die chronisch entzündlichen Darmerkrankungen Morbus Crohn und Colitis ulcerosa sowie die mikroskopischen Enterocolitiden. Die chronisch entzündlichen Darmerkrankungen sind bei manifestem Krankheitsstadium durch ihre systemische und endoskopisch-histologische Entzündungsaktivität (BKS-, CRP-, alpha-1- und alpha-2-Glykoproteinerhöhung, Thrombozytose) in der Regel mit einfachen Maßnahmen zu erkennen (1, 15, 17, 19, 25, 29, 33–37), während die mikroskopischen Enterocolitiden (kollagene oder lymphozytäre Enteritis bzw. Colitis) aufgrund des Fehlens systemischer Entzündungszeichen nur histologisch sicher identifiziert werden können.
Es ist daher obligat, bei Personen mit geschilderten gastrointestinalen Nahrungsmittelunverträglichkeiten, rezidivierenden Diarrhöen, Bauchschmerzen oder chronischen Magen-Darm-Problemen mittels transabdominellem Ultraschall, Endoskopie und Histologie, ggf. auch MRT-Sellink des Dünndarms, einen Befall durch Morbus Crohn oder Colitis ulcerosa bzw. mikroskopische Enterocolitiden auszuschließen. Gerade mikroskopische Enterocolitiden haben in den letzten beiden Jahrzehnten ebenso wie allergische Erkrankungen oder die eosinophile Ösophagitis (allergieartige, chronische Entzündung der Speiseröhre) eine deutliche Zunahme der Inzidenz und Frequenz erfahren, sodass heute differentialdiagnostisch danach gesucht werden muss, da gute Therapiemöglichkeiten bestehen (zum Beispiel Mesalazin, Budesonid [35, 37]).